Notable Research
Advancing regulatory variant effect prediction
with AlphaGenome
Technology Trend
학회뉴스
한양의대
DNA 서열로부터 그 효과를 추정하는 딥러닝 모델은 유전체의 조절을 예측하는 데 널리 이용되어 왔습니다. 하지만 기존의 방법들은 입력할 수 있는 서열의 길이와 예측 해상도가 반비례하여 성능에 한계가 있었습니다. 최근 제시된 통합 DNA 서열 기반 모델인 AlphaGenome은 1Mb의 DNA 서열을 입력하여 단일 염기 수준의 해상도로 다양한 유전체 변화를 예측할 수 있습니다.
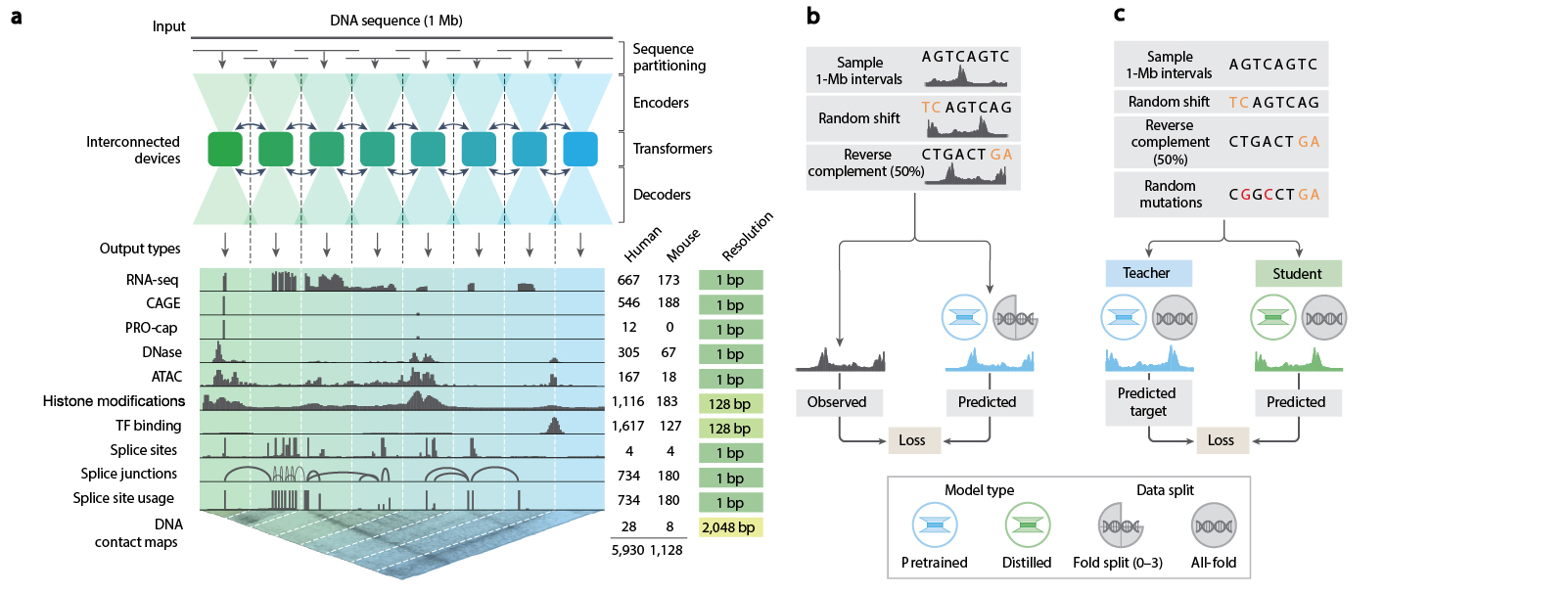 그림1
그림1
AlphaGenome 모델 개요
AlphaGenome은 인간 및 마우스 DNA를 학습하여 염기서열 기반으로 11개 범주의 유전체 변화를 예측합니다. 여기에는 유전자 발현(RNA-seq, CAGE, PRO-cap), 자세한 스플라이싱 유형(splice sites, splice site usage, splice junction), chromatin state(DNase, ATAC-seq, histon modification, transcription factor binding), chromatin contact map이 포함됩니다. 이러한 예측을 위해서는 1Mb의 DNA 서열을 입력하는데, 이 길이는 유전체에서 이루어지는 원거리 조절(distal regulation)을 상당 부분 포함하도록 설계된 것입니다. 예를 들어 99%(471개 중 465개)의 검증된 enhancer-gene pair가 1Mb 이내에 위치합니다(그림 1a).
AlphaGenome 모델은 두 단계의 과정을 통해 훈련되었습니다. 사전 학습 단계(그림 1b)에서는 관측 실험 데이터를 사용하여 두 유형의 모델을 생성했습니다. Fold-specific 모델은 4-fold crossvalidation 방식으로 학습되었으며, 참조 유전체의 3/4은 트레이닝에, 나머지 1/4은 검증과 테스트에 사용되었습니다. 또한 all-fold 모델은 모든 참조 유전체로 트레이닝 되었으며 두 번째 단계에서 teaching 모델로 활용되었습니다(그림 1c). 두 번째 단계에서 생성된 student 모델은 다양한 입력서열을 사용하여 all-fold teaching 모델의 예측을 재현하도록 학습되며 최종적으로 변이 효과 예측에 적합한 단일 모델을 생성합니다. 이러한 방식은 여러 독립적으로 학습된 모델을 결합하는 방법에 비해 대규모 변이 효과 예측에 더 효율적입니다.
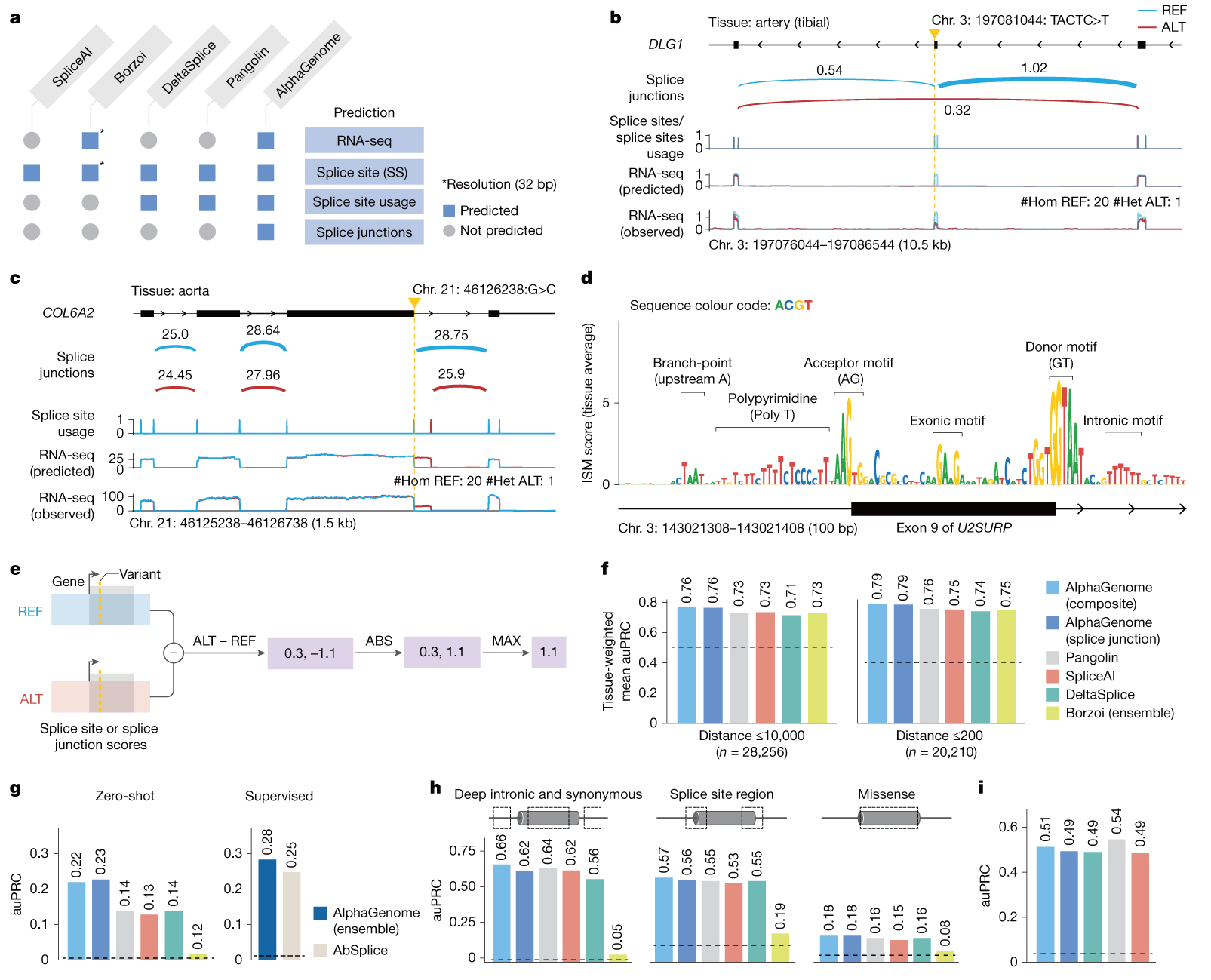 그림2
그림2
AlphaGenome의 splice variant effect 예측 및 유사 도구 간 비교
Splice variant의 효과를 예측하는데 있어 AlphaGenome은 세 가지 수준을 모델링하는데, 우선 주어진 염기가 splice donor 혹은 acceptor로 작용할 확률(splice site 예측), 잠재적인 splice site 중에서 경쟁적으로 선택될 확률(splice site usage), 그리고 특정한 인트론 예측(splice junction 예측)입니다. 여기에 더해 RNA-seq coverage 또한 예측함으로써 변이에 의한 splicing 효과를 좀 더 포괄적으로 이해할 수 있습니다(그림 2a). 기존에 잘 알려진 변이의 효과를 정확하게 예측할 뿐만 아니라(그림 2b, DLG1 gene 4-bp deletion; 영향을 받은 엑손의 splice site usage 감소, 소실된 엑손 경계의 junction 소실, 새로운 junction의 추정, 소실된 엑손의 RNA-seq coverage 감소), in-silico mutagenesis (ISM)로 생성한 모든 단일염기 변이 예측 분석에서도 splicing에 영향을 줄 수 있는 여러 가지 요인들을 밝혀냈습니다(그림 2d).
그 외에도 AlphaGenome은 gene expression, chromatin accessibility를 예측할 수 있으며 다양한 유전체 모달리티를 통합한(cross-modality) 변이의 효과 예측에 활용될 수 있습니다. 이러한 통합적인 변이 효과 예측은 복잡한 메커니즘을 가진 변이를 이해하는 데 도움을 주며, 유전체 전체의 조절서열인자(regulatory sequence element)를 분석하는 대규모 연구를 가능하게 합니다. 실험 연구에서는 AlphaGenome을 활용한 컴퓨터 기반 실험으로 신속하게 가설을 도출하고 실험실 실험의 우선순위를 정하는데 도움을 주며, 더 개선된 변이 효과 예측을 통해 희귀질환 진단 및 non-coding variant에 대한 판독 근거를 제공합니다. 또한 유전자 치료 목적의 anti-sense oligonucleotide 및 조직 특이적 enhancer와 같은 염기서열 설계에 활용할 수 있으며 DNA 서열로 훈련된 모델의 기능을 보완하는데도 이용할 수 있습니다.
그러나 여전히 몇 가지 제한점이 존재합니다. 우선, 100kb 이상 떨어진 원거리 조절 요소의 영향을 정확히 포착하기 위해 지속적인 개발이 필요합니다. 또한 AlphaGenome은 조직 특이적, 세포 유형 특이적 유전체의 특성을 상당히 잘 예측하지만, 세포 전반에 걸친 조직 특이적 패턴을 정확히 재현하고 특정 조건 하에 변이 효과를 예측하는 것은 여전히 어려운 부분입니다. 또한 훈련 데이터와 평가에 사용한 데이터가 단백질 코딩 유전자에 집중되어 있으므로 microRNA와 같은 non-coding 유전자를 통합할 수 있는 추가 연구가 필요합니다. 그리고 해당 모델은 인간과 마우스만 대상으로 훈련되었고, 평가 역시 인간 중심으로 이루어졌다는 한계가 있습니다. 마지막으로 AlphaGenome은 변이의 분자유전적 결과를 예측할 수는 있지만 복합형질분석에 적용은 제한적입니다. 이러한 표현형은 유전체 서열과 기능의 범위를 넘어서서 유전자의 기능, 발달 및 환경적 요인을 포함한 광범위한 생물학적인 이해와 유전자-질환 효과를 포함하는 영역이기 때문입니다.
요약하면, AlphaGenome은 조절 유전체(regulatory genome) 분석을 위한 통합적 모델입니다. DNA로부터 분자 기능과 변이의 효과를 예측하는 능력을 향상하고, 이를 통해 세포에서 이루어지는 복잡한 과정을 광범위하게 이해하는데 도움을 주는 도구라고 할 수 있습니다.
[References]
[1] Advancing regulatory variant effect prediction with AlphaGenome. Nature. 2026;649(8099):1206-1218.
