성균관의대
전장유전체분석(whole genome sequencing, WGS)의 도입으로 유전체 분석의 범위가 확대되고 다양한 유형의 유전체 변이를 검출할 수 있게 되었음에도 불구하고, WGS 기반 분석의 진단율은 여전히 약 30–40% 수준에 머물러 있다. 최근에는 이러한 미진단 사례의 상당 부분이 단백질을 직접 변화시키는 변이가 아니라 유전자 발현을 조절하는 비암호화(noncoding) 영역의 변이와 관련될 가능성이 제기되고 있다.
이 글에서는 비암호화 영역 중에서도 유전자 발현 조절에 관여하는 조절 서열을 중심으로, 이러한 요소의 특징과 이를 규명하기 위한 후성유전체 분석 방법, MPRA와 CRISPR 기반의 기능 분석, 그리고 최근의 인공지능 기반 예측 모델을 중심으로 비암호화 변이의 기능적 영향을 해석하기 위한 주요 접근법들을 살펴보고자 한다.
비암호화 영역에는 다양한 기능적 요소가 존재하며, 대표적으로 조절 서열(cis-regulatory element, CRE), intron, untranslated region (UTR), 그리고 다양한 noncoding RNA 영역 등이 포함된다. 이 중 CRE는 promoter, enhancer, silencer, insulator 등으로 구성되며, 전사인자(transcription factor, TF)와의 결합을 통해 유전자 발현을 정밀하게 조절하는 역할을 한다. 이러한 조절 서열에 변이가 존재하는 경우 TF와의 결합력, 염색질 접근성(chromatin accessibility), 또는 3차원 염색질 구조의 변화가 일어나 유전자 발현 수준에 영향을 줄 수 있다.
후성유전체 분석을 통한 조절 서열의 규명유전체 상에서 CRE의 위치와 기능을 파악하기 위해서는 다양한 후성유전체(epigenomic) 분석 기법이 활용된다. 염색질 접근성은 ATAC-seq 또는 DNase-seq를 통해 확인할 수 있으며, 특정 히스톤 변형(예: H3K27ac, H3K4me1 등)은 ChIP-seq 분석을 통해 enhancer 또는 promoter의 활성 상태를 추정하는 데 사용된다. 또한 전사인자 결합 위치 역시 ChIP-seq를 통해 규명할 수 있다. 이와 함께 염색질의 3차원 구조를 분석하는 3C(chromosome conformation capture) 기반 방법들(예: Hi-C, Capture Hi-C 등)은 enhancer와 promoter 사이의 물리적 상호작용을 파악하는 데 활용되며, 특정 조절 서열과 표적 유전자 간의 연결 관계를 이해하는 데 중요한 정보를 제공한다. 이러한 다양한 후성유전체 데이터를 통합하여 인간 유전체의 조절 서열을 체계적으로 정의하려는 노력이 진행되어 왔으며, 대표적인 예로 ENCODE 프로젝트를 들 수 있다.
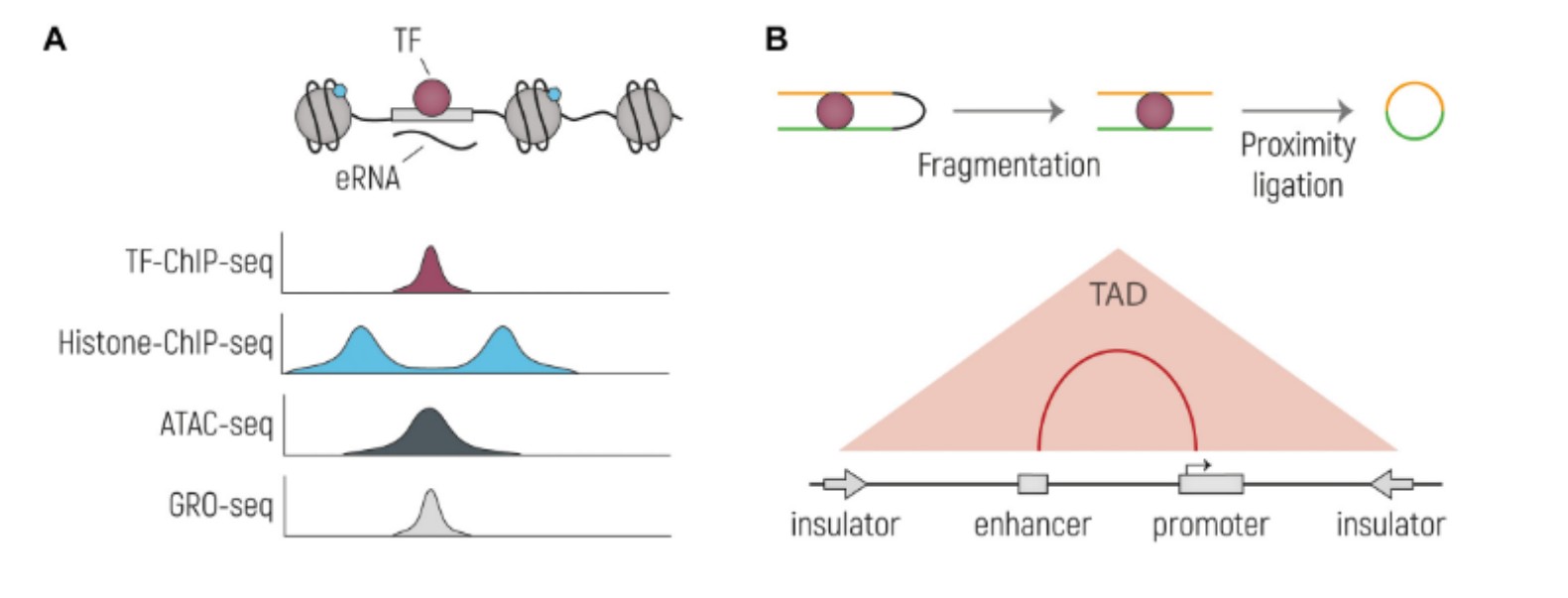 그림1
그림1
enhancer 서열과 그 표적 유전자를 규명하기 위한 주요 접근법의 개요
최근에는 비암호화 변이가 유전자 발현에 미치는 영향을 실험적으로 검증하기 위한 다양한 방법이 개발되어 왔으며, 그 대표적인 방법으로 massively parallel reporter assay (MPRA)를 들 수 있다. MPRA는 수천 개 이상의 후보 조절 서열 또는 해당 위치의 다양한 변이의 기능을 동시에 평가할 수 있는 highthroughput 분석 방법으로, 각 후보 조절 서열을 reporter gene 앞에 위치시키고, 각 서열에 고유한 DNA 바코드를 부여한다. 이후 RNA sequencing을 통해 reporter gene과 함께 발현된 바코드를 정량화함으로써 각각의 후보 서열이 유전자 발현에 미치는 영향을 동시에 측정할 수 있다. MPRA의 일종인 lentiMPRA는 후보 서열을 렌티바이러스 벡터를 이용해 세포의 유전체에 통합함으로써 chromatin context를 보다 유전체 환경에 가깝게 반영할 수 있다는 장점이 있다. 이러한 접근은 기존의 MPRA에서 후보 조절 서열이 episomal plasmid 형태로 존재한다는 한계를 일부 보완하며, 조절 서열의 활성을 보다 생리적인 환경에서 평가할 수 있게 한다.
CRISPR 기반 기능 분석과 Perturb-seq다만, MPRA를 통해 기능이 확인된 서열이라 하더라도 이는 reporter gene의 발현에 미치는 영향이 확인된 것이며, 해당 조절 서열이 실제 표적 유전자에 미치는 영향이 직접적으로 검증된 것은 아니다. 따라서 MPRA에서 발굴된 후보 조절 서열에 대해서는 추가적인 기능 검증이 필요하다. 최근에는 CRISPR 기반 기술을 활용하여 이러한 후보 서열의 기능을 세포 내에서 직접 검증하려는 연구가 활발히 진행되고 있다. 예를 들어 CRISPR interference (CRISPRi) 또는 CRISPR activation (CRISPRa)을 이용하여 특정 조절 서열의 활성을 억제하거나 활성화한 뒤 single-cell RNA sequencing을 수행하면, 해당 영역이 유전자 발현에 미치는 영향을 세포 수준에서 분석할 수 있다.
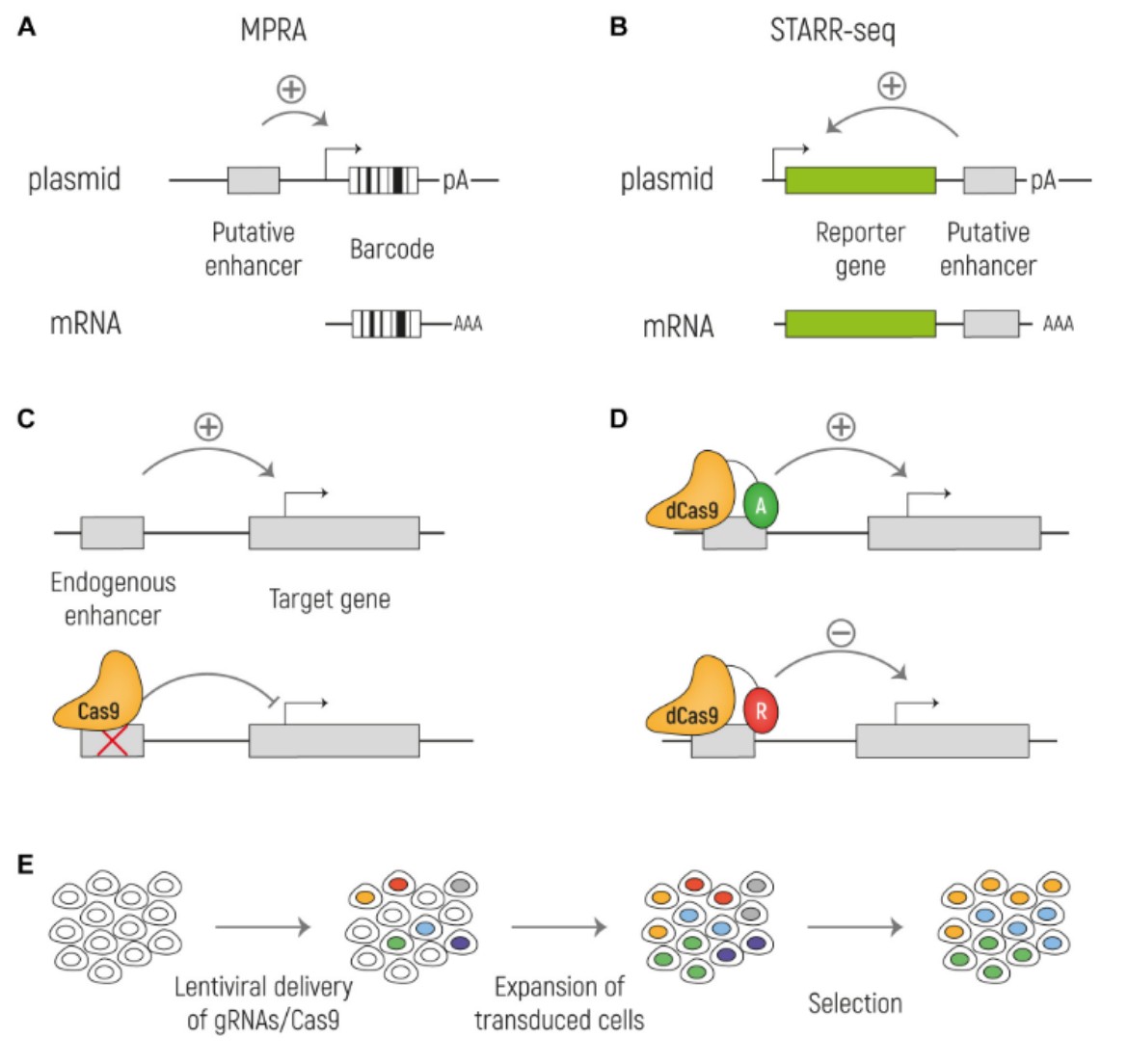 그림2
그림2
enhancer 기능을 검증하기 위한 주요 실험적 접근법의 개요
최근에는 인공지능 기반 모델을 통해 비암호화 변이의 기능적 영향을 예측하려는 시도들이 이루어지고 있다. 최근 발표된 AlphaGenome은 DNA 서열만을 입력으로 하여 다양한 유전자 조절 관련 결과를 예측하도록 설계된 딥러닝 모델이다. 이 모델은 전사인자 결합, 염색질 접근성, 히스톤 변형, RNA 발현, splicing 등 다양한 genomic signal을 동시에 예측할 수 있으며, 특정 변이가 이러한 분자적 신호에 어떤 영향을 주는지 in silico에서 예측할 수 있다. 이러한 인공지능 기반 접근법은 방대한 양의 비암호화 변이 중 실제로 기능적 영향을 가질 가능성이 높은 변이를 선별하는 데 도움을 줄 수 있으며, 이를 토대로 후속 실험 연구의 우선 순위를 설정하는데 도움이 될 수 있을 것으로 기대된다.
맺음말공공 기능유전체 데이터베이스 기반의 생물정보학적 주석(annotation), 인공지능 기반 기능 예측, 그리고 MPRA, CRISPR와 같은 실험적 검증을 함께 활용하면 변이의 기능적 영향을 보다 신뢰성 있게 평가할 수 있을 것으로 생각된다. 이를 통해 기존 WGS 분석으로도 원인을 규명하지 못했던 미진단 사례의 원인 변이를 밝히고, 유전 질환의 분자적 병태생리에 대한 이해를 넓힐 수 있을 것으로 기대된다.
[References]
1.Shlyueva, D., Stampfel, G. & Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;4:272-86.
2.Chatterjee, S. & Ahituv, N. Gene Regulatory Elements, Major Drivers of Human Disease. Annu Rev Genomics Hum Genet. 2017;18:45-63.
3.Perenthaler, E., Yousefi, S., Niggl, E. & Barakat, T.S. Beyond the Exome: The Non-coding Genome and Enhancers in Neurodevelopmental Disorders and Malformations of Cortical Development. Front Cell Neurosci. 2019;13:352.
4.Ellingford, J.M., Ahn, J.W., Bagnall, R.D., Baralle, D., Barton, S., Campbell, et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022;14:73.
5.Avsec, Z., Latysheva, N., Cheng, J., Novati, G., Taylor, K.R., Ward, et al. Advancing regulatory variant effect prediction with AlphaGenome. Nature. 2026;649:1206-1218.
